
CAR-T Therapy: How Can CRISPR Help?
Donors Cells, Immune System Evasion, AvenCell/Intellia/CRISPR Therapeutics

I wrote recently about CRISPR, specifically focusing on its applications in treating sickle-cell disease, beta thalassemia, and a newborn with severe liver disease. As a very brief summary, the CRISPR-Cas9 complex was initially discovered in bacteria as a way to protect against foreign intruders. When a bacteriophage that’s previously invaded injects itself into a bacteria, the Crispr-Cas9 complex recognizes the phage and neutralizes it with a double-stranded break in its DNA. The phage is recognized due to guideRNA, a transcribed slice of the phage DNA that was inserted into the bacteria’s CRISPR array after the first invasion. When the Cas9 enzyme finds a foreign DNA strand that’s complementary to the guideRNA, it initiates the double-stranded break.1

What makes CRISPR-Cas9 exciting for treating human disease is that we’re able to create synthetic gRNA, and so in theory can use the complex to affect whatever DNA sequence we like. Take sickle-cell disease, a blood disorder. We know there’s a sub-segment of those with sickle cell who never stop producing fetal hemoglobin. These patients either exhibit very mild symptoms or are entirely asymptomatic. We also know the gene responsible for repressing fetal-hemoglobin transcription. If I can design a gRNA strand complementary to this gene, the Cas9 will make a double-stranded break, and the human body’s imperfect DNA repair mechanism (non-homologous end-joining) will inactivate the gene in its efforts to rejoin the break. This is termed a gene-knockout approach, and is exactly what CRISPR Therapeutics/Vertex have done with Casgevy.
We can also modify the Crispr-Cas9 complex so that it alters a single nucleotide rather than makes a double-stranded break. An adenine base-editor turns an adenine base into a guanine, and a cytosine base-editor turns a cytosine into a thymine. This in turn changes an A-T base pair to a G-C one, and a C-G base pair to a T-A. An adenine base-editor was used in the case of newborn KJ in an effort to correct his CPS1 enzyme deficiency.
The potential applications of CRISPR don’t stop there (Intellia’s Annual Report is a fun place to look for where CRISPR could be used in the future. The company’s trying to tackle a lot!) CAR-T is an area where CRISPR may end up being especially useful. CAR-T, like other immunotherapies, is a way to harness the body’s immune system to more effectively fight off cancer. A patient’s own T-cells are removed, then modified in a lab to contain a chimeric antigen receptor (or CAR). Once transfused back into the patient, these chimeric antigen receptors will bind to antigens on cancerous cells, and so can more effectively fight off the cancer. In other words, the lab serves to train a patient’s T-cells to recognize the malignant cells and fight them off. For example, FDA CAR-T therapies often bind to CD19, a protein expressed in B-cells, and so worth targeting when treating B-cell cancers. There are currently 7 FDA approved CAR-T therapies, all of which are used for various blood cancers in cases where the cancer has returned after treatment or stopped responding to treatment.
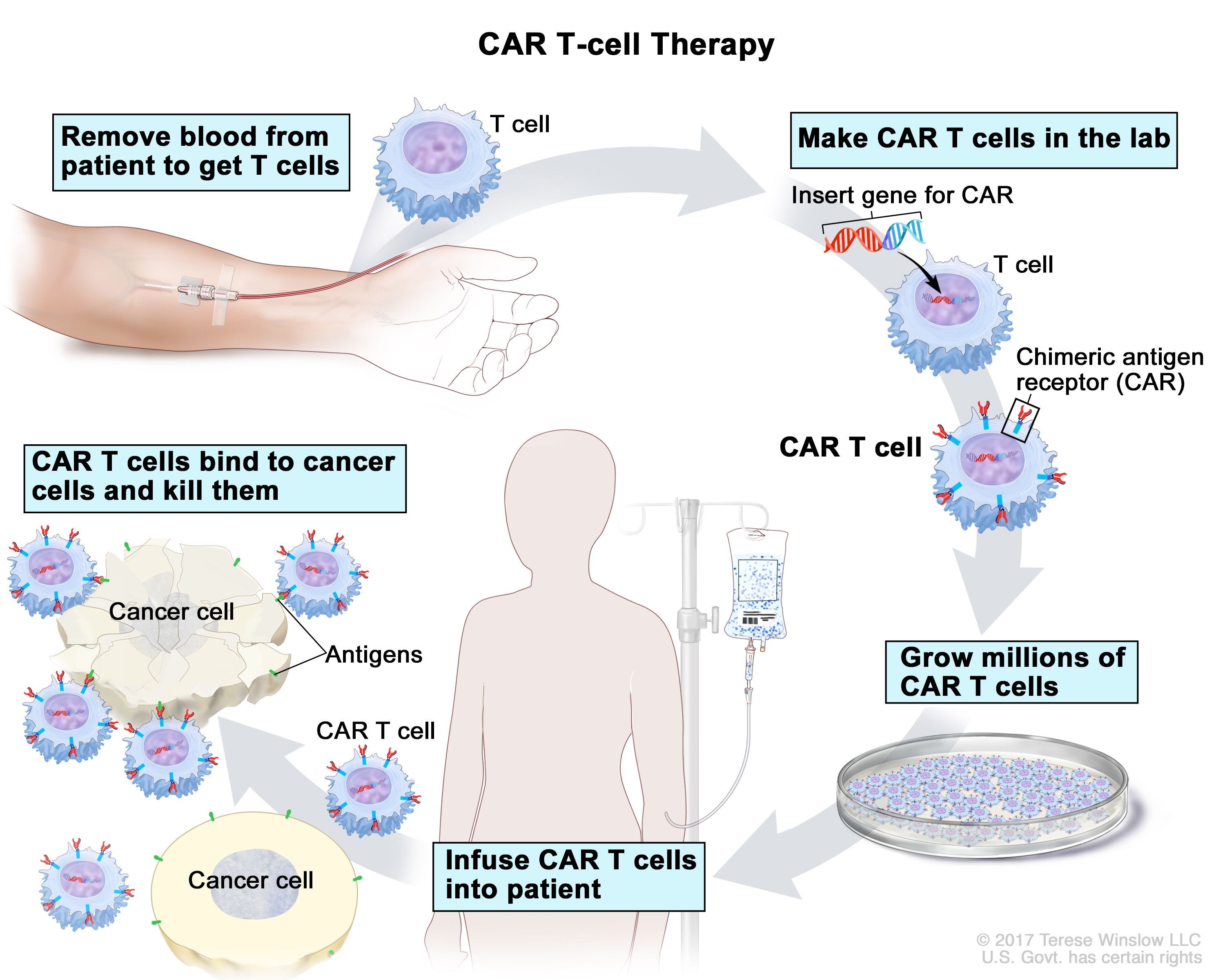
The seven FDA-approved treatments are all autologous, meaning they use a patient’s own T-cells rather than those of a donor. Autologous CAR-T therapies have several downsides. Modifying a patient’s own T-cells is far from an easy thing to do, and the process takes weeks to months, during which time the cancer can progress. The complexity of modifying these cells results in a costly treatment. Kymriah, the first FDA approved CAR-T treatment, is priced at $475,000; there isn’t much in the way of economies of scale when delivering bespoke therapies! The total cost is greater once one accounts for the costs of leukapheresis (the process of removing a patient’s white blood cells) and lymphodepletion therapy (a dose of chemotherapy the patient receives pre-infusion). Furthermore, an autologous approach requires that a patient’s T-cells are healthy enough to undergo modification. As one would expect with those who are very sick, this unfortunately isn’t always the case.
For the reasons given above, there’s a lot of interest in developing allogeneic (donor derived), rather than autologous, CAR-T treatments. There are real benefits to an allogeneic approach: T-cells don’t need to be harvested from a sick patient, the patient doesn’t have to wait around for treatment, there are economics of scale in the manufacturing process, and patients with non-optimal T-cells can still be treated. Currently, however, this approach poses issues on both the donor and patient side:
Graft-versus-host disease – the donated T-cells see the recipient’s T-cells as a threat. This is due to a mismatch in human leukocyte antigens (HLAs), proteins on human cells that let the immune system know which cells are native/foreign. The donor T-cells detect they’re in foreign territory, and so begin attacking the patient rather than helping him/her. The donor cells’ T-cell receptors (TCR) are responsible for launching this attack.
Rejection via host T cells - The host -cells aren’t not pleased to have the donor cells there. Again, this is due to mismatches between donor and acceptor HLAs. Two types of host T-cells attack these donor cells: CD8 and CD4.
Rejection via host NK cells – NK cells, or natural killer cells, are another type of white blood cell that attacks cells it deems harmful. Host NK cells also attack the donor cells.
Leveraging CRISPR-Cas9 is a potential avenue to solve these issues. Companies differ in their approaches to mitigate host T-cell and host NK cell rejection, but the CRISPR approach to prevent graft-versus-host disease has been fairly uniform. The ability to create synthetic gRNA means scientists can decide where to insert the chimeric-antigen receptor, something that isn’t possible with typical CAR-T transduction methods. With CRISPR, the CAR gene can be inserted into the TRAC locus, which is the section of the T-cell responsible for TCR expression. Placing the CAR gene here disrupts the TRAC locus, thus preventing TCR expression and graft-versus-host disease.
Avoiding rejection via host T-cells and NK-cells is more complex. As mentioned above, CD8 and CD4 host T-cells both attack the donor cells. CD8 cells are activated when they detect foreign HLA I molecules; CD4 cells are activated when they detect foreign HLA II molecules. Knowing this, the obvious solution would be to use CRISPR-Cas9 to knockout HLA I/II expression in the donor cells. Unfortunately, natural killer cells spring into action when they detect cells with little to no HLA-I expression. Knocking out HLA-I might prevent CD8 cells springing to life, but at the cost of activating NK cells!
The easiest method, at least initially, to solve for the issue of host immune response is lymphodepletion, which is a standard part of the autologous CAR-T process anyway. Lymphodepletion gets rid of a large number of patient lymphocytes (T-cells, B-cells, NK-cells), thus giving the incoming CAR-T cells plenty of room to proliferate and fight off the cancer. This in turn increases the odds of CAR-T cell persistence, which has been continually linked to better outcomes. Of course, this means the patient’s immune system is in a comprised state, which is why infections are a common side effect of CAR-T.
As said above, this works as an initial way to limit host immune response. The patient’s immune system will come back, and at that point T-cells and NK cells will get to work once they notice HLA mismatches in the donor T-cells. This response limits CAR-T persistence, and thus the effect of the therapy. One option here is to up the dose of chemotherapy and thus keep the immune system suppressed for longer, but that in turn further increases the risk of patient infection.
An effective treatment approach that leverages allogeneic CAR-T, then, is all about figuring out how to get those CAR-T cells to endure in patients. This problem is in some ways made more acute by disrupting the TRAC locus: this step prevents graft-versus-host disease but actually reduces donor T-cell persistence. CRISPR Therapeutics and Intellia Therapeutics are both working on allogeneic CAR-T therapeutics that leverage CRISPR technology, but have taken different steps to get there.
Intellia has focused predominantly on evading immune detection. They knockout HLA Class II genes, thus evading rejection by host CD4 T-cells. As mentioned above, knocking out HLA Class I genes isn’t as simple, because that results in activated host NK cells. Intellia’s workaround here is to knockout only HLA-A, which is part of HLA Class I. They then keep HLA-B, C, and E, and search for T-cells donors whose HLA-B and C alleles are matches to that of the patient. This should prevent host NK Cell and CD8 T-cell rejection. Moreover, it would significantly simplify the donor matching process: management estimates 20 donors would cover 80% of the US population.
CRISPR Therapeutics takes a blended approach of avoiding immune detection and knocking out genes that reduce T-cell persistence in other ways. They knockout the entirety of HLA Class I, and additionally knockout the genes that code for Regnase-1 and TGFBR2. Knocking out Regnase-1 should increase T-cell persistence and antitumor immunity; knocking out TGFBR2 should prevent CAR-T cell exhaustion (when T-cells stop functioning as they should). CRISPR Therapeutics’ decision to knockout the whole of HLA Class I and not touch Class II means there will be an immune response from host CD4 T-cells and host NK cells, but they’re betting that knocking out Regnase-1 and TGFBR2 will make up for that.
Developing allogeneic CAR-T treatments, whether using CRISPR or not, has been a tough place to be over the past few years. Enthusiasm for gene/cell therapies has dropped dramatically since Covid, leading to frequent layoffs and slashing of programs in development. Most of the companies in the space are in phase I trials with market caps of sub 300mm USD (Caribou, Cellectis, Celyad, Fate, Imugene). Allogene has a large B-cell lymphoma treatment in phase 2 trials, but its market cap also sits at ~264mm. Not all of them leverage CRISPR. Allogene and Cellectis both use TALEN, another gene editing tool that leverages mechanisms initially found in bacteria; Celyad doesn’t use gene editing at all given concerns about off target effects; Imugene uses yet another gene-editing tool (ARCUS) discovered by Precision Biosciences.
Sana Biotechnology only has phase I candidates, but has done a fantastic job raising money and a promising, non CAR-T, diabetes type 1 treatment in the pipeline. Gracell was acquired for a billion by AstraZeneca, but the acquisition announcement emphasized the company’s autologous CAR-T manufacturing capabilities rather than its allogeneic programs. Roche spent a billion to acquire Poseida Therapeutics in late 2024, and in that case indicated real interest in the company’s CRISPR edited allogeneic CAR-T candidates. CRISPR Therapeutics and Legend Biotech are doing fine, but both have FDA approved therapies in other areas and do much more than just allogeneic CAR-T!2 AvenCell, a private company working on both autologous and allogeneic candidates, looks quite interesting. The company licensed Intellia’s platform for its allogeneic candidates, and raised over 100mm in late 2024, a tough time to be raising capital when you’re not an AI business! Blackstone Life Sciences was its founding investor back in 2021, which is either another data point on the style drift that occurred in the Covid era (the fund typically invests in much later-stage drugs) or an indication the company’s working on something with a relatively high likelihood of success.
Disclaimer: The information in this post is not intended to be and does not constitute investment or financial advice. You should not make any decision based on the information presented without conducting independent due diligence
Legend is another company pursuing a non-gene editing approach to modifying donor cells.