
Chemoproteomics: Scorpion/Antares, Vividion Tx, Frontier Medicines
Or, covalent inhibitors pt 2.

I wrote recently on covalent inhibitors, focusing on these drugs’ mechanism of action and why binding affinity is an insufficient measure of their potential effectiveness. I also briefly discussed the toxicity/off-target effect concerns that, historically, meant preclinical candidates known to be covalent were frequently killed before entering the clinic. We have plenty of FDA-approved covalent drugs (aspirin and penicillin among them!), but their covalent mechanism often wasn’t realized pre-approval.
The general perception of covalent compounds has changed meaningfully over the past decade, so much so that covalent-first approaches to drug discovery are now able to raise large sums of VC money (see here) or merit an acquisition by big pharma (see here and here).
Covalent-first approaches to drug discovery generally refer to companies using covalent chemical probes and proteomics to find druggable targets on a protein. The whole raison d’etre of such companies is to develop covalent molecules, and they tend to refer to themselves as ‘chemoproteomic platforms.’
The most frequently used chemoproteomics method in the covalent context emerged from activity-based protein profiling (ABPP), a method historically used to assess enzyme activity within the proteome. The general pitch behind ABPP is this:
Proteomics is an effective way to measure protein expression levels. However, it doesn’t tell you much about protein activity. As was put well here, “Protein presence does not imply protein functionality.” As a result, proteomics is limited when one is trying to understand whether an enzyme is in an active or inactive state. It is here that ABPP enters the picture. In ABPP, a chemical probe is used that binds specifically with an enzyme’s active site. This chemical probe contains an electrophilic warhead that will react with a nucleophilic residue within the active site, thus forming a covalent bond. In other words, it acts in the same way as a covalent drug. Mass spectrometry is then used to analyze the results.
The chemical probe used varies based on the enzyme one is analyzing, but chemical probes targeting cysteine residues become especially relevant in the drug discovery/development context. Cysteine is highly nucleophilic, relatively rare, but also often present within or close to binding sites.1 Consequently, ABPP targeting cysteines can be very helpful for finding previously undiscovered binding pockets. The methodology of ABPP remains the same, but the goal shifts from using the cysteine to analyze enzyme activity.
Chemoproteomics is not the only method one can use to find previously unknown binding sites, but its covalent approach does offer a unique advantage. Namely, its real promise is that it should be more effective at figuring out ways to drug proteins today considered ‘undruggable.’
This promise is worth harping on a bit because it’s different from what a lot of recently approved or late-stage trial covalent drugs do. It’s also different from what many recently approved or late-stage trial non-covalent drugs targeting novel binding sites do. Take Osimertinib, an EGFR inhibitor used in types of non-small cell lung cancer. Osimertinib binds covalently, and is more selective for mutant EGFR over wild-type. This is a real improvement over previous generations of EGFR inhibitors. However, it doesn’t target an ‘undruggable’ protein. Lilly recently acquired Scorpion Therapeutics’ mutant-selective PIK3CA inhibitor, STX-478. Scorpion (whose other assets have been spun out into a new company Antares), is a chemoproteomics company, and does have candidates in the pipeline targeting undruggable proteins. However, its acquired asset, STX-478, acts covalently but isn’t targeting a protein we weren’t already able to target.2
The same can be said of Relay’s PIK3CA inhibitor. Relay isn’t a covalent company, but does focus on developing drugs that bind to previously undiscovered binding sites.3 Again, this can have real value! But it doesn’t necessarily result in drugging a target that was thought to be undruggable. The same can also be said for Terns’ lead asset/Novartis’ Scemblix for CML: both bind to an interesting allosteric pocket that delivers improved tolerability/potentially improved efficacy, but they’re not drugging a new target.
I don’t mean to suggest that the above isn’t unimportant work. That said, where chemoproteomics really gets exciting is when it comes to these targets that we haven’t been able to drug yet. Adagrasib/Sotorasib are the quintessential examples. These were the first drugs to actually succeed at targeting RAS, which we’ve known was a cancer driver since the 80s but haven’t been able to do anything about. Using disulfide tethering, another chemoproteomics method also leveraging covalent bonds, Kevin Shokat and his team were able to:4
1) Identify a previously unknown RAS binding pocket (termed Switch-II).
2) Design a compound to bind to that target
The covalent component specifically is vital here. As said in my first note, this bond is what enables getting beyond subpar binding affinity that precludes using a non-covalent molecule. To be clear, not all previously undiscovered binding pockets will require a covalently acting drug (as Relay’s PIK3CA inhibitor demonstrates), but there are undiscovered binding pockets where only a covalent drug will be able to effectively bind to it. Put differently, it’s possible a tool like molecular dynamics or cryo-EM could’ve surfaced the previously unknown RAS Switch II binding pocket, but that pocket still wasn’t targetable without a covalent drug. Because chemoproteomics screening uses the same core mechanism that the eventual drug will, it increases the odds that the pocket you find will be truly actionable.
Companies like Vividion Therapeutics, Frontier Medicines, and Scorpion (now Antares) are trying to systematize the rough approach used by Shokat. We know the most about Vividion, and its S1 (pre Bayer’s acquisition) gives a helpful window into the general approach these companies take to drug discovery/drug development. Broadly speaking, it’s split into mapping and screening:
“The mapping step determines the maximum set of solvent-exposed cysteines that can participate in a covalent reaction across the entire proteome. We accomplish this by using a non-selective, cysteine specific chemical probe that has been designed to maximize both cysteine reactivity and mass-spectrometric detectability. This initial map, which has been elucidated in the absence of any other cysteine reactive small molecules, serves as the reference set of potential cysteine sites for covalent small molecule engagement”5
In short, the goal here is to find all the cysteines that could potentially be bound to with a covalent small molecule. This mapping chemical probe could never be turned into an actual drug: its broad reactivity means it would have extensive off-target effects.
The ‘screening step’, then, is the attempt to screen potential compounds for selectivity against the specific protein of interest. Such a step is especially important for covalent drugs, where off-target binding poses more threats than off-target binding from traditional small molecules given the irreversible bond. In the context of this screening step, Vividion’s S1 reads:
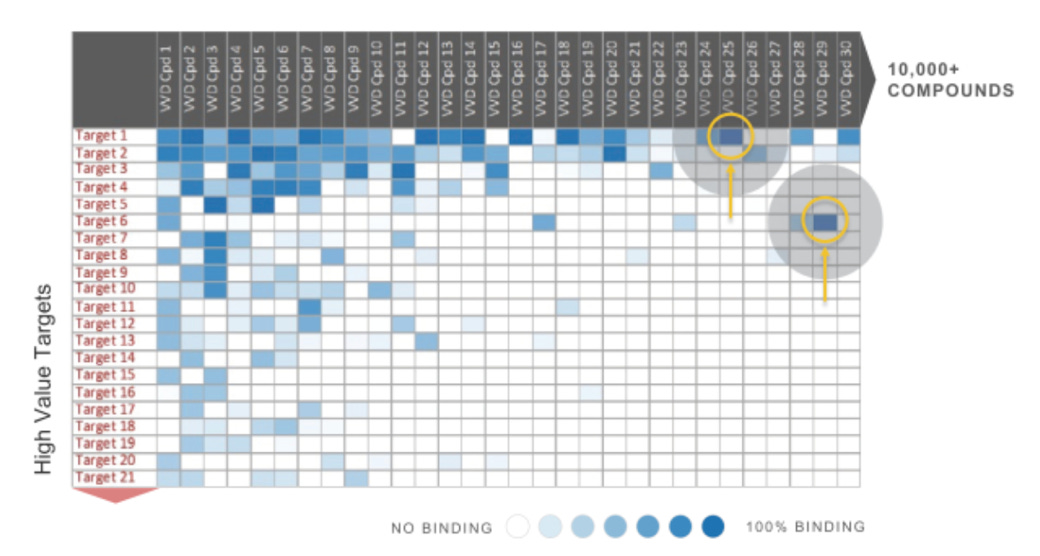
“To illustrate the value of selectivity information, in the example below there are many compounds that potently inhibit target #1. However compound #25 does so with compelling selectivity, whereas many other compounds display off-target binding.”6
I think the most exciting stuff going on in the chemoproteomics world relates to transcription factors, which are proteins that regulate gene expression (by either activating or repressing initial DNA to RNA transcription). Much like RAS, aberrant transcription factors are known to be cancer drivers, but are often standard ‘undruggable’ targets with no clear binding pockets. Some transcription factors are more straightforward, and in the cases where we can target them it tends to work quite well. Estrogen receptors are transcription factors often overactive in breast cancer. Fulvestrant is used to treat certain advanced breast cancers and works by selectively degrading these estrogen receptors.
Antares is collaborating with AstraZeneca on the transcription factor front, and Frontier has a candidate targeting re-activation (rather than inhibition) of p53 via the cysteine mutation at the protein’s 220th codon (as well as a collaboration with Abbvie targeting an undisclosed transcription factor). c-MYC is another transcription factor that people like Daniel Nomura are very excited about potentially targeting. Like p53 mutations and RAS mutations, c-MYC has been known to be a cancer driver since the 80s. Unfortunately, c-MYC is intrinsically disordered (for any philosophers reading I don’t mean this in Aquinas’ sense), meaning that rather than taking on a stable 3D shape it flips rapidly between multiple different conformations. It’s hard to find a binding pocket for a protein that doesn’t want to sit still!
There are two final points worth making here. The first is the downside of these covalent approaches to undruggable proteins is that they’re only able to target very specific mutations. Adagrasib/Sotorasib were real innovations, but their targeting of the G12C mutation means they basically don’t have applications to PDAC, where Ras mutations are present in over 90% of all cases. In contrast, Revolution Medicine’s molecular glue approach is so exciting because it enables pan-RAS inhibition. Frontier’s p53 work is very exciting, and I hope they succeed. That said, while p53 is mutated/deleted in ~50% of all cancers, p53 mutations that occur at the Y220C mark are only present in 1-2% of cancers. Again, this isn’t meant to disparage the covalent approach; I think it’s fascinating, and I also hope that the field succeeds in extending beyond cysteines. The downside, though, is that it’s hard to see a world where a covalent drug can act upon multiple forms of a mutated protein in the way that a molecular glue can.
The second point to make is that a covalent approach is not necessarily required when developing therapeutics that target novel binding sites. This is a common feature of any biotechnology company’s S1 filing, but one could come away from the Vividion S1 thinking that PROTACs, allosteric inhibitors/activators, and protein-protein interaction modulators only work if there’s a covalent bond involved. Many PROTACs act non-covalently, Relay’s PIK3CA allosteric inhibitor acts non-covalently, and RevMed’s lead candidate inhibits an ‘undruggable’ target without using a covalent bond. Ibrutinib may have put covalent inhibitors back on the map, but there are now even BTK inhibitors that act non-covalently. Covalent drugs are a promising approach for widening the aperture of small molecules, but fortunately are one of a few options!
Disclaimer: The information in this post is not intended to be and does not constitute investment or financial advice. You should not make any decision based on the information presented without conducting independent due diligence.
Its relatively rare nature also means one can be less worried about off-target effects.
Again, this isn’t to say STX-478 won’t represent a real step forward from existing PIK3CA inhibitors. Developing STX-478 is also a smart business decision. Develop a much improved version of an inhibitor that we know has demonstrated clinical efficacy, then use the proceeds from that drug to develop candidates for truly ‘undruggable’ targets.
I would highly recommend reading about the Relay Therapeutics D.E. Shaw Research collaboration. a
Disulfide tethering is a chemoproteomic approach that leverages reversible covalent probes rather than irreversible ones. As one would guess from the name, these chemical probes form disulfide bonds with ligandable cysteines. Interestingly, one of its developers was Daniel Erlanson, now Chief Innovation Officer at Frontier Medicines. It’s fallen a bit out of favor compared to ABPP because it does require that you decide your target ahead of time. While Shokat didn’t know about the switch-II binding pocket, he did know he was trying to target the RAS G12C mutation. In contrast, ABPP allows for surveying of the entire proteome without deciding where to focus.
Pg 126, Vividion S1.
Pg 127, Vividion S1