
Covalent Inhibitors Part 1
RAS, For Blood and Money, Vividion/Scorpion/Frontier

This research service has spent a good amount of time recently discussing ‘undruggable’ targets, whether that’s Aktis and its miniprotein platform or Revolution Medicines and its pan-RAS inhibitor. I’ve also spent time on therapies targeting unique binding sites for already druggable proteins. Here, the goal is to improve upon existing drugs’ side effect profiles by minimizing inhibition of non-target proteins, often by binding allosterically.
Covalent drugs straddle these two worlds. They can be used to target proteins previously thought undruggable, and to create more selective/tolerable versions of existing inhibitors. Such drugs have experienced a surge in popularity in recent years. Ibrutinib and Acalabrutinib, both covalent BTK inhibitors, were the subjects of For Blood and Money1 Sotorasib and Adagrasib, covalent inhibitors used to treat KRAS G12C-mutated NSCLC and colon cancers, were the first approved drugs that managed to bind to RAS. We’ve known RAS to be a cancer driver since the 80s, so it’s fairly big deal that covalent drugs were the ones to crack the code after decades of failed efforts!
Unlike traditional small molecule drugs, which function via non-covalent interactions, covalent ones contain an electrophilic ‘warhead.’ Once in position (which, importantly, is achieved non-covalently), this warhead is able to accept electrons from its nucleophilic counterpart (often cysteine) and form a covalent bond. There are technically reversible and irreversible covalent inhibitors, but for the purposes of this note we’ll focus only on irreversible. Irreversible is a bit of a misleading term, and in reality means the protein will be inhibited by the drug until it’s resynthesized (which could take 24 hrs plus). This is, however, very different from standard small molecules, where inhibition occurs for minutes to single digit hours.
Traditional, non-covalent, small molecules present real challenges when it comes to targeting ‘undruggable’ proteins that play a role in disease. These proteins tend to be characterized by flat surfaces with no obvious binding sites (think of trying to grab onto a flat, slippery, cliff face rather than a beginner bouldering hand hold). That’s not an environment where small molecules tend to thrive: the lack of clear binding sites means an interaction requires a far more extensive number of non-covalent bonds. That can be achieved with a much larger protein, but not with a molecule that’s ~100x smaller.
Covalent small molecules have an advantage here, namely that they’re not completely reliant upon binding affinity. This is unique compared to these other approaches for targeting ‘undruggable’ proteins. The thesis behind Aktis’ approach is that its miniproteins are large enough to generate a sufficient number of non-covalent interactions to bind to a flat protein. Consequently, their S1 emphasizes the sub-nanomolar/nanomolar binding affinity of its pipeline candidates. Revolution Medicines’ chief innovation is around molecular glues, which again act non-covalently!
Things are a little different when it comes to covalent compounds. These compounds operate in two steps:
1) Like traditional small molecules, the compound first binds to the protein via non-covalent bonds.
2) Next, the small molecule’s warhead forms a covalent bond with a closeby nucleophile.
One way to conceptualize these steps is to think about effectively shutting the front door on a windy day. Step one is closing the door. This puts the door in the correct position, but at some point it’ll get blown back open. So it is for regular small molecules, where the aim isn’t extended inhibition. However, if the goal is to keep the door closed overnight one needs to actually turn the lock. So it is with a covalent small molecule that’s trying to inhibit a protein for an extended period of time.
Binding affinity is still very important here. Without it, the molecule can’t get in position to form a covalent bond. However, the maximum potential rate of covalent bond formation (denoted as kinact) is also very important. kinact tells you the speed with which a covalent bond forms with the target protein assuming all of the target protein has formed the initial non-covalent complex.2 Taken together, kinact/KI describes the overall efficiency of covalent bond formation for these covalent inhibitors.
In other words, measuring binding affinity alone doesn’t give an accurate view into how well a covalent compound may be performing. This is well-illustrated in an example given by DrugHunter:3
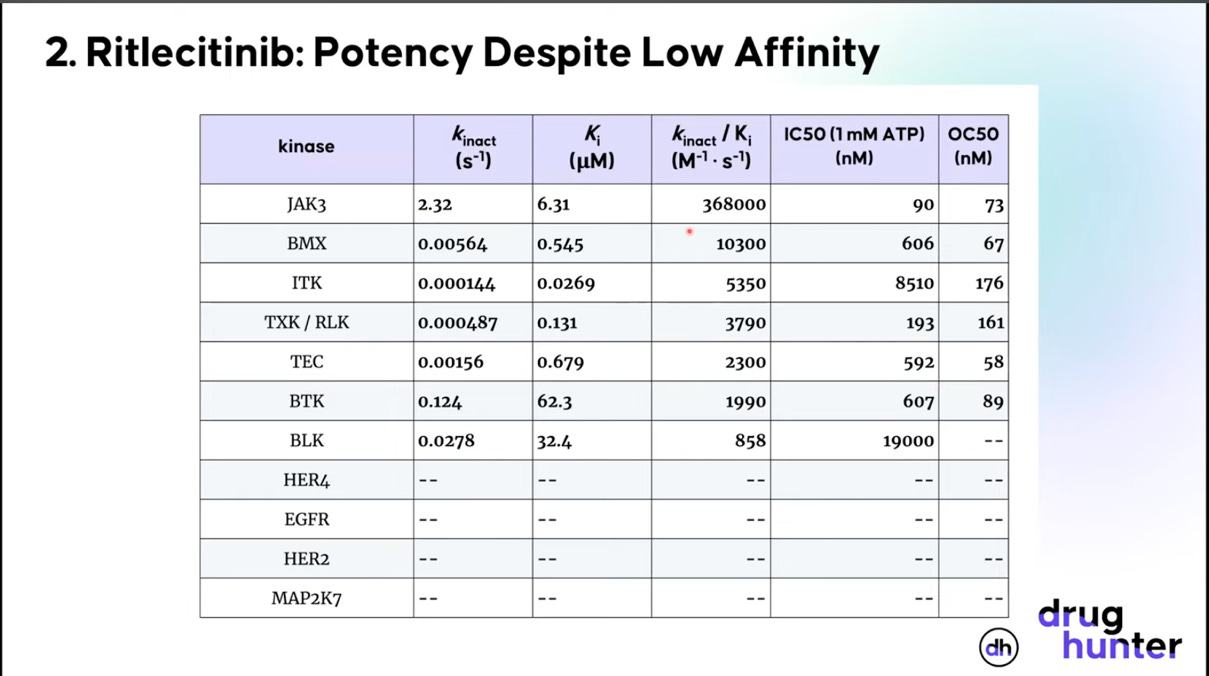
Ritlecinitib (Litfulo) is a covalent JAK3/TEC kinase family inhibitor, developed by Pfizer, for the treatment of alopecia areata, an autoimmune condition causing hair loss. The goal of the compound was to selectively inhibit JAK3, a task it doesn’t look very good at doing based on Ki alone. When you look at kinact and kinact/Ki, however, things look much more promising. The general takeaway is to be wary of a covalent drug developer spending too much time discussing binding affinity alone.45
The covalent nature of these compounds additionally means one can worry less about the drug’s pharmacokinetics. For non-covalent small molecules, tracking its plasma concentration (Cp) over time is critical for determining when a next dose might be needed. The lower the plasma concentration, the less drug there is inhibiting the target protein. For covalent compounds this isn’t the case. Plasma concentration matters initially, but only until the warhead reacts with its protein target. Once that occurs, the target remains inhibited until re-synthesis. In other words, Cp stops correlating with drug effect.
Historically, development of covalent molecules was shunned given concerns about toxicity and off-target effects. Covalent drugs can lead to hapten formation, where the drug covalently binds to a protein and the resulting complex causes a massive immune response. This isn’t something one has to worry about when a molecule can’t bind covalently! Moreover, off-target effects become a bigger issue when covalent bonds are involved: inadvertently inhibiting a non-target protein for 24 hours plus is a bigger concern than inhibiting it for only a few minutes. That’s not to say we weren’t using covalent drugs: Penicillin and Aspirin act covalently, but their covalent nature wasn’t known at the time of approval. Put another way, the challenge with trying to develop a drug known to be covalent is that it would be discarded/deprioritized in favor of other compounds.6 This is no longer the case. Revolution Medicines’ lead candidate may act only non-covalently, but the company has earlier-stage candidates that throw in a covalent bond on top of the molecular glue. The 2021/2022 approval of Sotorasib/Adagrasib means covalent drugs are now seen as having real potential for hitting ‘undruggable’ proteins. There are even companies like Vividion Tx, Frontier Medicines, and Scorpion Tx, whose entire thesis is that chemoproteomics can find protein binding pockets that are promising targets for covalent compounds. Vividion was acquired by Bayer for $1.5 billion, and Lilly will spend potentially $2.5 billion acquiring only one of Scorpion’s candidates (the other assets have been spun-out into a new entity called Antares).7 Such big pharma interest in covalent first approaches would’ve been hard to imagine 15 years ago.
Disclaimer: The information in this post is not intended to be and does not constitute investment or financial advice. You should not make any decision based on the information presented without conducting independent due diligence.
Ibrutinib was also the drug that put Robert Duggan on the map as a biotech executive. Duggan’s Wikipedia, as well as this WSJ profile on him, are very much worth reading. Not many people have run a bakery business and a few different biotech companies.
I’d recommend the following article to those interested in learning more about this second-order constant.
KI and Ki, though similar, are technically not the same metric. From this paper: “KI and Ki are not interchangeable in much the same way that KM and Kd for the substrate are not interchangeable. The main distinction between KI and Ki is that the former includes the contribution of kinact. Strictly, KI can approximate Ki only when koff is much larger than kinact, which is often the case.”
That’s very much not to say that binding affinity doesn’t matter. Osimertinib is a covalent EGFR inhibitor that improves upon earlier EGFR inhibitors by more selectively binding to mutant-EGFR. Part of this improved selectivity is because the compound has improved binding affinity for mutant-EGFR over wild-type.
This is exactly what happens in For Blood and Money when Merck acquires Oss as part of Schering-Plough