
Revolution Medicines: The Transition from RAS(OFF) to RAS(ON) $rvmd
Molecular glues, off-target effects, covalent bonds

One of the things that arose in my writing on Aktis is the challenges researchers/companies have faced historically in targeting protein-protein interactions (PPIs). To review, not all proteins have well-defined binding sites. The enzyme-substrate interactions one learns about in high school are unique in their lock-and-key nature. Often proteins interact with each other primarily through many weak intermolecular contacts, not because protein A is a perfect fit for protein B’s binding site.
The importance of targeting PPIs is perhaps perfectly illustrated through RAS proteins. RAS proteins have been known to drive many types of cancers since the 1980s, and yet for decades there wasn’t anything that could be done to inhibit these proteins’ activity. There was no obvious binding site that could be leveraged to inhibit RAS function. RAS exists primarily in two states: active and inactive, or RAS(ON) and RAS(OFF). In its active state, RAS is bound to GTP; in its inactive state RAS is bound to GDP. To further complicate drug design efforts, RAS binds to GTP and GDP with picomolar affinity, which makes those proteins incredibly hard to displace with an alternative.
In healthy cells, RAS proteins are essential for cellular proliferation, differentiation, and survival. Typically, RAS exists primarily in the off state; if you looked at the ratio of inactive to active RAS proteins in the average cell, RAS(OFF) would far outweigh RAS(ON). This dynamic is often flipped in cancerous cells, where increased RAS(ON) activation allows the cancerous cells to flourish at the expense of their host. It’s worth emphasizing that these RAS mutations are known to be driving the cancerous growth; we’re not discussing a passenger mutation. RAS exists in three different isoforms: HRAS, NRAS, and KRAS. Most conversations center around KRAS, as it’s a more prevalent oncogenic driver. RAS mutations are most commonly present in lung (~30% of all cases), colon (~40% of all cases), and pancreatic cancers (>90% of all cases), so most of the discussion centers around treating those diseases:
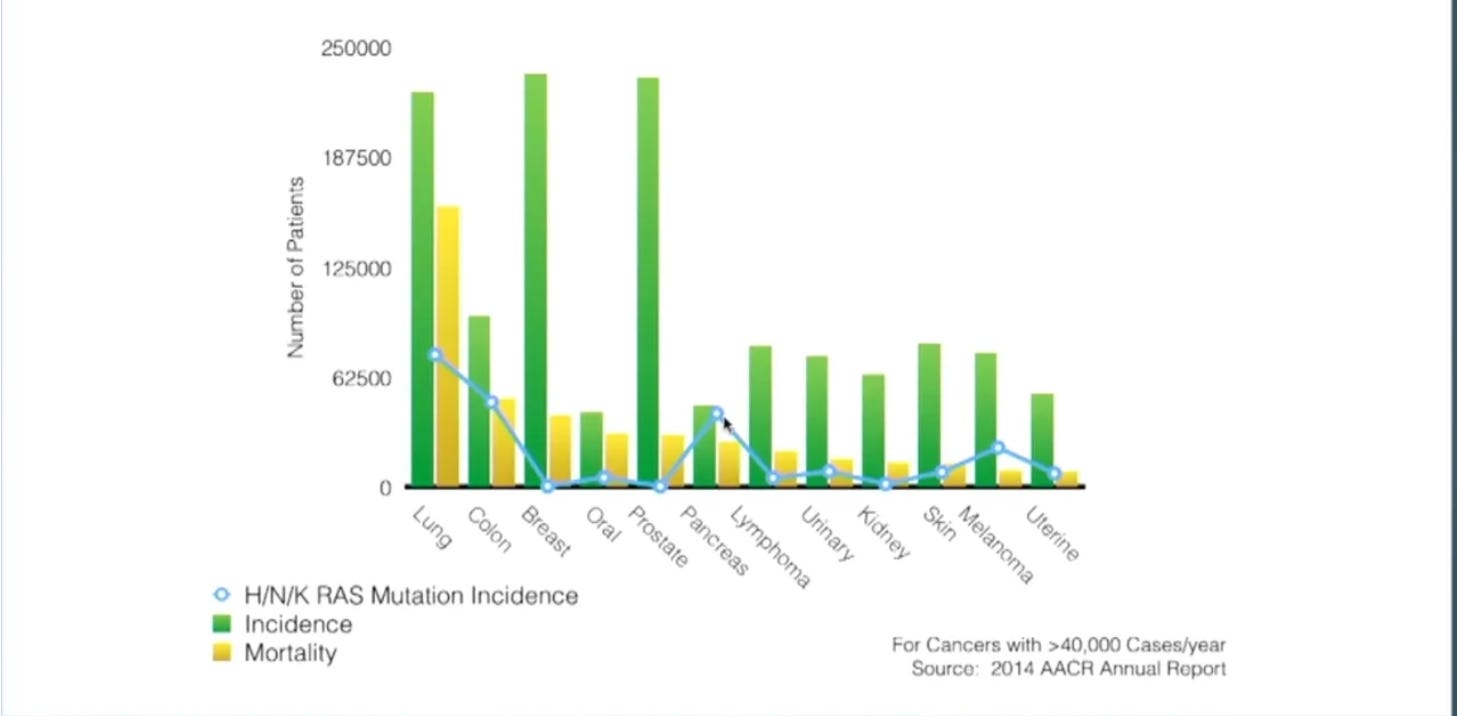
The first approved RAS-targeting therapeutics were Sotorasib and Adagrasib, approved in 2021 and 2022, respectively. Both were initially approved for previously treated patients with KRAS G12C-mutated non-small cell lung cancer (NSCLC). Both are now also approved as part of combination therapies to treat KRAS G12C-mutated colon cancer.
Sotorasib/Adagrasib represent true innovation and are based on the work of Kevan Shokat out of UCSF. The KRAS G12C targeting piece is essential for understanding how these drugs work. For patients with this mutation, the 12th codon of the KRAS gene is altered. This alteration turns what should be the amino acid glycine into the amino acid cysteine. This cysteine mutation, specifically, is actually very useful when it comes to drug development. It’s highly nucleophilic, or in other words eager to form covalent bonds. Sotorasib/Adagrasib takes advantage of this nucleophilicity, forming a covalent bond with the cysteine mutation when RAS is in an inactive state.1 These compounds don’t try to disrupt the actual RAS-GDP complex; such an approach would be incredibly challenging given the strong binding affinity between RAS-GDP. Instead, the covalent bond with the cysteine mutation prevents RAS from switching back into its active state, and so should positively affect the ratio of RAS(ON) to RAS(OFF) proteins in the cell.
Sotorasib/Adagrasib’s binding to the cysteine residue makes them highly selective. This cysteine mutation only occurs in mutant KRAS proteins, so there’s little risk of affecting healthy KRAS or off-target effects.2 There are downsides, however:
Both drugs work initially, but treatment resistance does eventually occur.
Given than RAS(ON) is what’s driving cancer growth, Sotorasib/Adagrasib’s RAS(OFF) binding isn’t ideal. Lowering the number of mutant RAS(ON) proteins in circulation is a good thing, but you’d still ideally be directly targeting RAS in its active state.
These drugs work because of the cysteine amino acid specifically; again, cysteine is highly motivated to form covalent bonds. It’s much harder to design drugs targeting a different amino acid mutation, even if it’s still on that 12th codon. The G12D mutation is actually a more common driver of cancer, but it’s not as nucleophilic.
The KRAS G12C mutation is only present in 14% of NSCLC cases and 3% of colon cancer cases. The G12C mutation is even less prevalent in pancreatic ductal adenocarcinoma (1-2%), the poster child of RAS-driven cancers.
This last point illustrates a drawback of the mutation-specific approach generally: it means one’s limited to developing drugs in a mutation-by-mutation manner. In an ideal world, there’d be a drug that’s just as effective treating patients with a G12C mutation as it is at treating those with G12D, G13C mutations, or others.
Historically, there was good reason to think such an approach wasn’t feasible. The challenge with broadening the selectivity of a RAS inhibitor is that this increases the odds you’ll end up affecting the body’s healthy RAS cells. In other words, you’re going in the opposite direction from the general trend in cancer treatment. Pluvicto targets the PSMA antigen because it’s heavily expressed in prostate cancer cells and minimally expressed in healthy ones. RAS multi-selective inhibitors target RAS proteins because they’re cancer drivers, but with the trade-off that you risk affecting the healthy RAS proteins present throughout the body. In other words, it’s a little bit like traditional chemotherapy.
It is here that Revolution Medicines enters the picture. The company went public in February 2020, and at the time was best described as a very interesting small molecule company going after a variety of targets, one of which was RAS. Its focus has narrowed since then, and Revolution Medicines now describes itself as “a clinical stage precision oncology company developing novel targeted therapies for RAS-addicted cancers.”3 Its two most mature candidates are:
Daraxonrasib – a multi-selective RAS inhibitor currently in registrational trials for PDAC (the most common form of pancreatic cancer) and NSCLC. This pan-RAS inhibitor makes use of only non-covalent bonds.
Zoldonrasib – a G12D-selecitve RAS inhibitor in registrational trials for PDAC. Like existing RAS inhibitors, Zoldonrasib makes use of a covalent bond to increase selectivity.
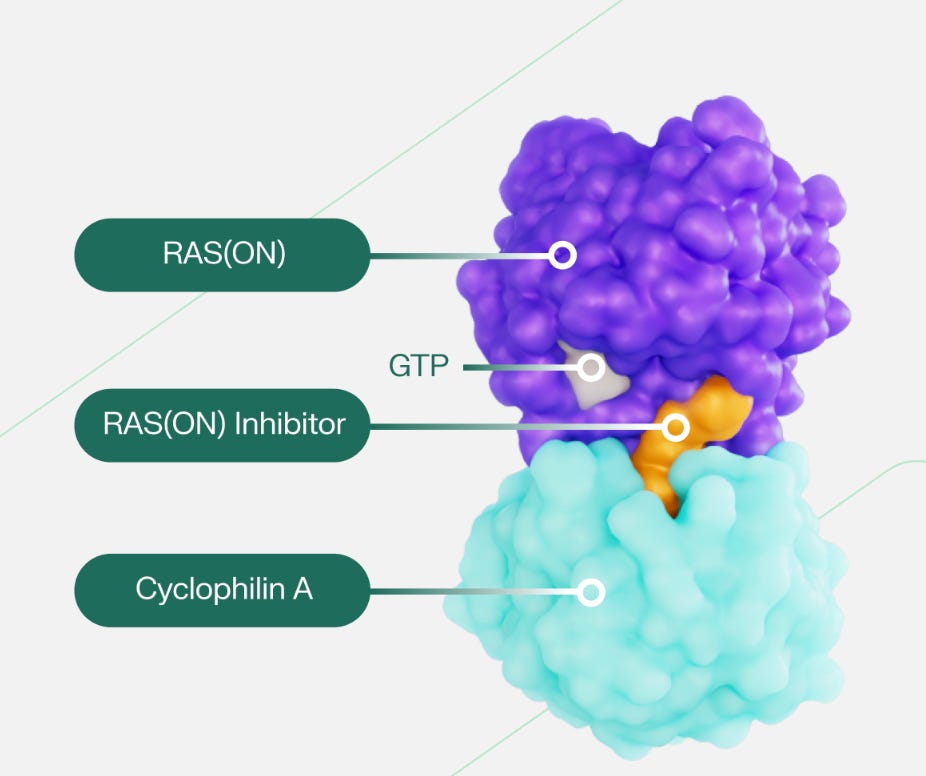
Critically, Revolution’s RAS inhibitors bind to RAS(ON) rather than RAS(OFF), and so more directly attack what’s causing cancer proliferation. The company’s candidates are all small molecules, but small molecules that act in quite a unique way. Daraxonrasib and Zoldonrasib are considered molecular glues, which means they’re small molecules used to affect protein-protein interactions.4 The small molecule binds to one protein, which alters that protein’s binding surface. This altered surface enables an interaction with another protein that wasn’t previously possible. These glues have actually been used clinically for a while, even if it wasn’t clear that was the mechanism of action: take Cyclosporin A, which has been approved for use in organ transplants since the early 1980s. Cyclosporin A enters the cells and binds to Cyclophilin (most commonly cyclophilin A). This binding alters the shape of Cyclophilin, which now enables the binary complex to bind to Calcineurin. Inhibiting Calcneurin function dampens the immune system, thus preventing organ transplant rejection.
Revolution Medicines’ candidates act by blocking RAS(ON) from initiating its deleterious downstream effects. Like Cyclosporin, the inhibitor enters the cell and binds to Cyclophilin A. This binding alters the shape of Cyp(A), which enables this complex to now bind to RAS(ON) and inhibit its function5 This protein-protein interaction is, in the case of Daraxonrasib, comprised entirely of non-covalent interactions. That’s what one would expect when trying to modulate a PPI: for protein surfaces with no obvious binding site, one is reliant upon many weak intermolecular contacts.6 It’s worth emphasizing that Cyp(A) and the small molecule are both necessary for this RAS inhibition to occur; neither is sufficient by itself. Revolution refers to the RAS(ON)-Cyp(A)-small molecule block as a tri-complex.
Both Daraxonrasib and Zoldonrasib are exciting therapeutic candidates for two reasons:
They’re potentially able to target RAS(ON)
They can do so for a cancer type, PDAC, that has very few promising treatments, is often metastatic at the time of diagnosis, and comes with a RAS mutation an overwhelming amount of the time. Revolution’s decision to focus on its G12D inhibitor over its G12C candidate is smart: G12D mutations are present in ~40% of PDAC cancers.7
Said differently, there’s a very good reason why the company received a National Priority Voucher for Daraxonrasib. The uniquely fascinating part of Daraxonrasib, specifically, is that its side-effect profile wasn’t intolerably severe. This is the case even though it’s inhibiting mutant and wild-type RAS proteins. Dr. Kenneth Olive at the Olive Lab has a theory for why this is the case:
“First, RMC-7977 does not inhibit RAS continuously in tissues. Over the course of 24 hours after a dose, the RAS pathway is fully inhibited within a few hours, but it is back to normal after 12-24 hours. We think this allows normal tissues to recover and get enough of the normal RAS function to continue to be healthy. At the same time, we found that tumor cells react violently to the sudden loss of RAS signaling, with many of them dying within a few hours of the first dose (and more dying after each subsequent dose). Tumor cells seem to be addicted to RAS signaling whereas normal healthy cells can get by for a bit without it. This makes pan-RAS inhibition “tumor-selective”8
I think it’s fair to say excitement about Revolution Medicines is baked into the stock price. The company needs a lot to go right beyond treating second-line metastatic PDAC to grow into a 20 billion dollar valuation. 61,000 patients in the US are diagnosed with PDAC every year. 52% of those are diagnosed with metastatic. Of that 52%, ~56% actually get treatment. Of that 56%, ~46% receive second-line treatment. That puts you at 8200 patients every year. If you assume 100% patient penetration (a bit unlikely!), that these patients stay on Daraxonrasib for 8.5 months (the median PFS survival period in the company’s released results), and that Daraxonrasib is priced at ~20k per month (similar to Sotorasib/Adagrasib), that gets you ~1.3 billion USD in peak U.S. sales each year.9 There’s of course potential for Daraxonrasib on the NSCLC side (and on the colon cancer/breast cancer side), but in general that’s a more crowded space. Furthermore, Daraxonrasib does unfortunately seem to work only for a period of time before the cancer progresses again; this isn’t a cure.
That’s not necessarily to say the valuation doesn’t make sense. The 2L PDAC data really has been quite promising so far, and could represent a big step up from the current standard of care. Additionally, there’s plenty more in the late-stage pipeline. If Daraxonrasib is approved for 1L metastatic PDAC (whether as a combo or solo treatment), things start to get really interesting. Moreover, Revolution Medicines really is the company that’s figured out a potential way to disrupt RAS in its active form. I would guess that part of the valuation premium here is down to how innovative the approach is, even compared to other developers of RAS-targeting treatments. A pan-RAS inhibitor is also the archetypal pipeline in a product: RAS is known to be implicated across plenty of different cancers (even if, thankfully, standards of care for other cancers tend to be better than for pancreatic). Finally, Baker Bros is a large shareholder, and historically they have a bit of an eye for promising approaches to cancer! (see Seagen)

Disclaimer: The information in this post is not intended to be and does not constitute investment or financial advice. You should not make any decision based on the information presented without conducting independent due diligence.
These small molecules are both beyond the rule of 5 compounds given their high molecular weights.
This is a pertinent risk for some of the Huntington Disease treatments currently in clinical trials.
Pg 3, 2026 Annual Report.
Molecular glues are often talked about today in the context of protein degradation, where they’re leveraged to facilitate the actual breakdown of cancer driving proteins within a cell.
As with Sotorasib/Adagrasib, the goal is not to somehow displace the strong binding affinity between RAS-GDP or RAS-GTP.
I do think the bouldering analogy is a helpful one here. A beginner climber pursuing a challenging route has to rely upon many weak limb connections to the wall. This is quite unlike an easy course where one’s hand can comfortably grab onto a hold and support one’s entire weight.
Sotorasib/Adagrasib likely would have started with G12D if they could have, but, again, their approach only worked because of the cysteine amino acid specifically.
Source here. RMC-7977 is technically not the exact same as Daraxonrasib but it molecularly very similar. From the same piece: “RMC-6236 [Daraxonrasib] is an investigational drug that is being developed by RevMed for use in humans. It is currently in clinical trials. RMC-7977 is a preclinical tool compound. RevMed makes this agent widely available to scientists so that they can carry out research on pan-RAS inhibition in different cancers without impacting the clinical development of their investigational agent. The two agents are closely matched in terms of molecular structure, pharmacology, and mechanism of action. Lessons from RMC-7977 should generally be applicable to RMC-6236.”
I’m assuming median PFS roughly matches median duration as treatment, which is what you see for Sotorasib/Adagrasib. I’m also assuming that Daraxonrasib is covered for all those with PDAC because RAS mutations are just so prevalent there (as we’ve seen with Padcev & bladder cancer).