
PROTACs: Protein Degradation for 'Undruggable' Targets
bRo5 molecules, Arvinas/Kymera, Further PK/PD Decoupling

Most annual report/S1 filings I’ve been reading of late have a similar pitch:
1) 80-85% of protein targets are undruggable with conventional small molecule/biologics methods
2) We have a novel approach that will enable us to drug these targets (whether that be molecular glues, covalent small molecules, or otherwise)
3) There are actually already FDA approved drugs that act via this mechanism; we just didn’t know it at the time of approval. (Aspirin/penicillin act covalently, Cyclosporin A is a molecular glue, Lenalidomide is a molecular glue degrader)
4) We are systematizing this approach and so should be able to develop therapeutics for clinically validated but currently undruggable targets (the best example here is Revolution Medicine’s Daraxonrasib. RAS has been known as a cancer driver since the 80s; Revolution Medicine’s molecular glue approach just actually made RAS broadly druggable)
PROTAC (proteolysis targeting chimera) companies fit the above description well.1 These companies take advantage of a natural biological process. Proteins, once synthesized, don’t exist forever. Once a protein has served its purpose (or become misfolded/damaged), it’s then degraded by the proteasome, the cell’s protein garbage disposal system. Historically we haven’t had drugs that were technically PROTACs, but we have had some drugs that take advantage of this innate degradation machinery. Fulvestrant, a breast cancer drug approved in 2002, acts by deforming estrogen receptors, which in turn means these receptors are degraded by the proteasome.
Importantly, the proteasome needs a way to recognize which proteins require degradation, and this is where ubiquitination comes in. Ubiquitin is a small protein that, when added to another protein in sufficient quantities, acts as a flashing light to let the proteasome know said protein needs to be degraded. The ubiquitination process is facilitated by 3 enzymes: E1, E2, and E3. Ubiquitin is activated by E1, transferred over to E2, and then the E3 ligase helps E2 transfer the ubiquitin over to the target protein (via a covalent bond to a lysine residue on the target).2
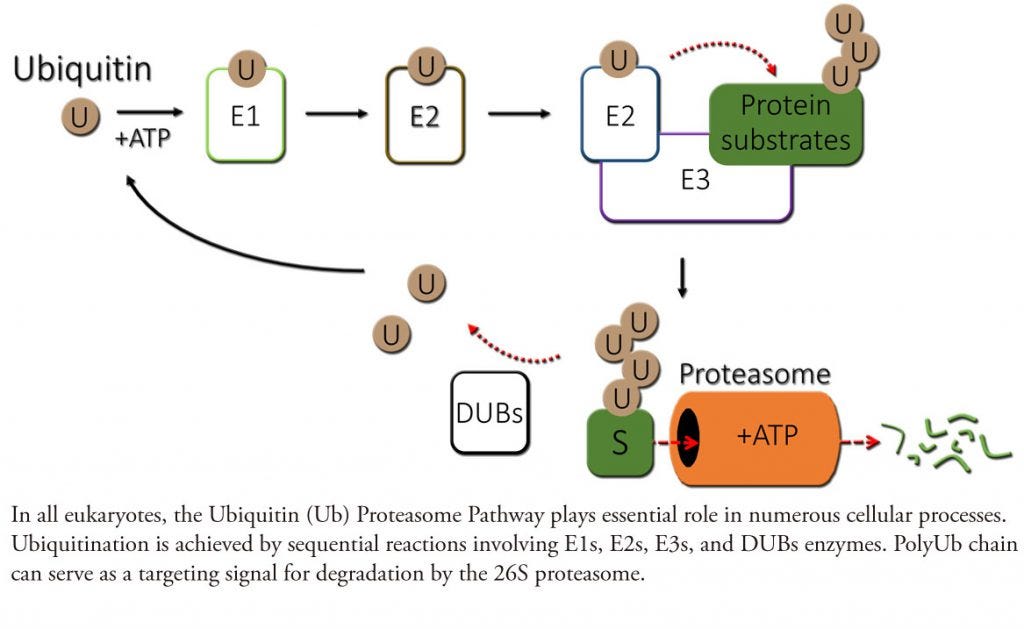
Approaches that leverage this innate degradation system are an attractive way to target proteins previously considered undruggable, partially because it gives drug developers some leeway in binding site selection. With standard small molecule drugs, the aim is to affect a target’s function in some way (whether by preventing or encouraging it). Consequently, one needs to either bind to an enzyme’s active site or to an allosteric site that will in turn affect the active site. (I went into this a bit in my note on Terns, but one can think of an allosteric site as similar to a boot on a car. One isn’t directly inhibiting the engine running, but the car is now undrivable). A traditional small molecule that binds to an enzyme at a non-functional binding site doesn’t accomplish anything, as the protein’s function is not affected. The same is true with covalent small-molecule approaches. Chemoproteomics may help you find a previously unidentified binding site, but that binding site still needs to affect the target’s function!3 With PROTACs, however, things change. The structure of a PROTAC typically consists of two separate small molecules connected by a linker. One small molecule binds to the target protein, the second small molecule connects to the E3 ligase, and this forms a ternary complex between the PROTAC, E3 ligase, and target protein. This encourages polyubiquitination and subsequent degradation of the target. The PROTAC dual small-molecule structure is described as a ‘heterobifunctional degrader.’
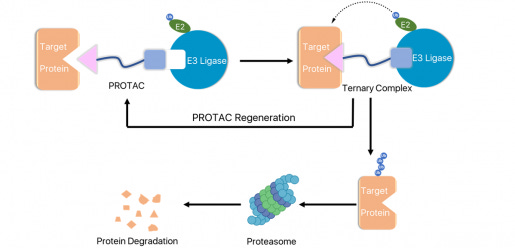
With PROTACs, then, the goal is only to bind to the target protein in some manner; one doesn’t necessarily need to bind to the protein in such a way that its function is inhibited. Instead, one only needs to bind to the protein in enough of a way to encourage a protein-protein interaction between the E3 ligase and target protein. While you still need a sufficient amount of binding affinity, expanding addressable binding sites beyond only the functional ones is a real advantage! There’s no need to inhibit a protein’s function if you can instead get the proteasome to degrade it.
As one might guess, this is very useful when it comes to affecting previously ‘undruggable’ targets. It’s also very handy in cases where a protein acts partially in ways that we do know how to drug and partially in ways we do not. IRAK4, a compelling target for inflammatory diseases and certain blood cancers, is a good example. The challenge is that it acts both as a traditional kinase (typically druggable) and as a scaffolding protein (typically undruggable). Resultantly, blocking only kinase function won’t necessarily do enough to affect the protein overall.
Like covalent drugs, PROTAC’s mechanism of action is such that binding affinity takes on a lower level of importance than for traditional small molecules. It’s still a relevant consideration, but favorable protein-protein interactions between the E3 ligase and target protein (in other words, the stability of the ternary complex) are even more so. A PROTAC with strong binding affinity but that can’t form a sufficiently stable ternary complex isn’t going to get anywhere. Metrics often used in PROTAC analysis are:
DC50 – the concentration of PROTAC required to degrade 50% of the target. As with typical binding affinity, a lower concentration of drug is better.
Dmax - the maximum level of target protein degradation that can be achieved with a PROTAC.
Again like covalent drugs, PROTACs are also a class of therapeutic where pharmacokinetics take on a differing level of importance. With covalent drugs, this is because the target is still inhibited even after the small molecule has exited the system (and will stay inhibited until the protein is re-synthesized). With PROTACs things are similar, as the protein has been degraded and so definitionally will not be functioning (nor present!) until resynthesized. Unlike typical small molecules, the drug’s plasma concentration isn’t going to track with the drug’s efficacy.
There’s an additional level of differentiation with PROTACs, in that they act ‘catalytically.’ Once a PROTAC has facilitated polyubiquitination of one target protein, it then goes back and does the same to another, and then another, and so on until it exits the body. This should mean one can use a lower dose than the equivalent, non-catalytic, traditional small molecule. This should in turn mean that, all else equal, PROTACs have a more benign side-effect profile.
There are downsides with PROTACS, most notably the ‘hook effect’ and these molecules’ large size. The hook effect refers to a phenomenon where a PROTAC’s efficacy can actually diminish the higher one takes the dose. In other words, once dosing reaches a certain point Dmax paradoxically starts to decrease. When there’s too much of a heterobifunctional degrader these molecules end up binding individually to the target protein and E3 ligases. Rather than one heterobifunctional degrader binding to first the target protein and then to the E3 ligase (or vice versa), heterobifunctional degrader A binds to the target protein, degrader B binds to the E3 ligase, and then one gets no ternary complex formation. This of course presents challenges when one wants to take the dose up to try and achieve higher efficacy.

PROTACs’ large size stems from the fact that they’re really two small molecules joined together with a linker. As a result, these PROTACs have molecular weights above 500 Da, violating part of Lipinski’s rule of 5 for oral small molecule design. This is an issue because typically small molecules with molecular weights above 500 Da also have polar surface areas that are too large. Small molecules are a trade-off between polarity and lipophilicity: too polar and the drug will have a hard time crossing cell-membranes/getting out of the GI tract in the first place, too-lipophilic and the drug’s solubility won’t be any good. Larger molecules often have the too polar issue: it’s not all that helpful to design a PROTAC for a hard to target transcription factor if one can’t even get into the cell! There’s a lot of work being done to figure out appropriate parameters for making ‘Beyond Rule of 5’ small molecules work, but it’s quite early days. To be clear, we do have some FDA approved small molecules larger than 500 Da, so the task is to figure out why these larger molecules escape traditional Rule of 5 parameters while others do not. This video with the Director of Global Discovery Chemistry at Novartis argues that we’re basically not nuanced enough in calculating polar surface area, which explains why we see some bRo5 compounds with polar surface areas that should be too high working effectively.
Kymera is probably the most notable PROTAC/targeted protein degradation public company with its STAT6 degrader: people tend to get quite excited about effective oral drugs in atopic derm that potentially won’t have a black box warning. Arvinas (which trades below cash & equivalents) is also doing some very interesting work in the PROTAC space. The company does actually have an FDA approved PROTAC drug for treating a subset of breast cancer patients, but it ended up working in a much narrower patient population than hoped (ER+/HER2- advanced/metastatic patients with an ESR1 mutation who have tried CDK 4/6 inhibitors and endocrine therapy, rather than ER+/HER2- advanced/metastatic patients who have tried CDK 4/6 inhibitors and endocrine therapy, irrespective of ESR1 mutation). That said, Arvinas is doing some fascinating things on the brain-penetrant PROTAC front. Small molecules have even stricter polar-surface area requirements when it comes to crossing the blood-brain barrier, so it’s pretty wild that a small molecule with a molecular weight of 800 Da+ may be able to do so.
Disclaimer: The information in this post is not intended to be and does not constitute investment or financial advice. You should not make any decision based on the information presented without conducting independent due diligence.
I’m focusing this note on PROTAC degraders specifically, rather than molecular glue degraders.
It’s not the PROTAC itself that is necessarily acting covalently (although some in development do). It’s a covalent bond between the target protein and an ubiquitin.
Chemoproteomics can also be helpful in the PROTACs context, because there may be a non-functional binding site identified that can only be grabbed onto via a covalent bond.